반응형
1. 미국 의료기기 규제 기관 소개
- 식품의약국 (Food and Drug Administration, FDA) 미국 보건 복지부의 산하기관으로 소비자 보호기관의 역할을 하는 국민보건법의 주무부처이다. 사람과 동물을 위한 의약품, 백신, 생물의약품의 사용 및 의료기기에 대한 안전성과 효과성 확보를 통해 국민의 건강을 보호하는 것을 주요 목적으로 한다. 1906 년 6 월 30 일에 창립되었으며, 본부는 메릴랜드 실버 스프링 (Silver Spring, MD) 에 위치해 있고, 부처 설립자는 테오도르 루즈벨트, 하비 워싱턴 윌리 (Theodore Roosevelt, Harvey Washington Wiley) 등 이다.
- 식품의약국은 9 개의 센터 및 13 개의 실 조직으로 구성 아래 링크를 클릭하면 식품의약국의 조직 구조와 산하 9개의 센터 및 13개의 실로 구성 되어 있는 조직도를 볼 수 있다.
[ 그림 1 ] 미국 FDA 조직도
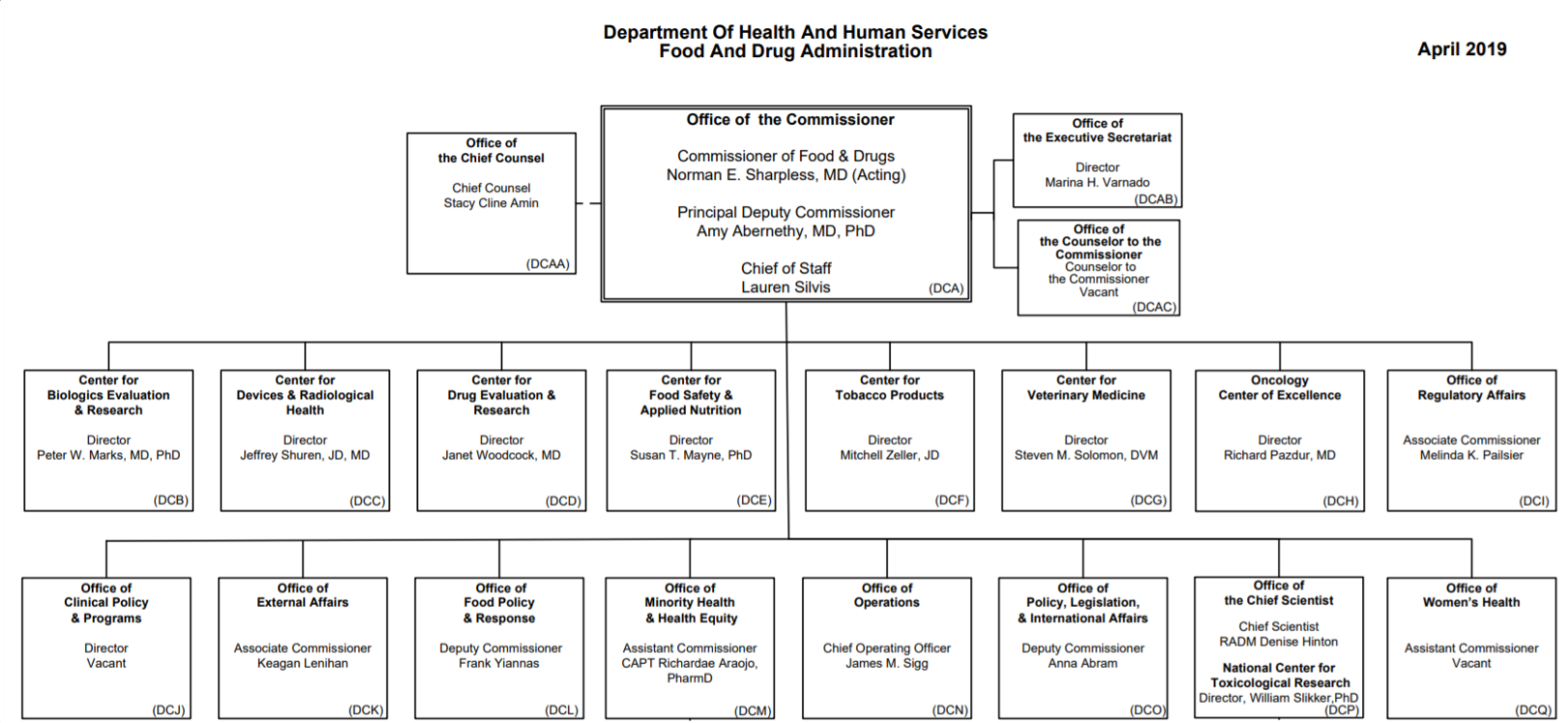
2. 식품의약국 산하 센터 (9개)
- Centerfor Biologics Evaluation and Research CBER(생물의약품평가연구센터)는 공공 건강 서비스 법령 (Public Health Service Act) 및 연방 식품, 의약, 및 화장품 법령(Food, Drug and Cosmetic Act)등에 따라 생물학 제재에 관한 사항을 규정 및 관리하는 센터이다.
- Centerfor Devices and Radiological Health4) CDRH(의료기기및방사능건강센터)는 환자와 의료인이 안전하고, 효과적이며 우수한 품질의 의료기기와 방사능 관련 기기 인허가를 심사 및 규제하는 센터이다.
- Centerfor Drug Evaluation and Research5) CDER(의약품평가연구센터)는 안전하고 효과적인 의약품 개발을 통해 공공의 건강을 증진하는 역할을 하는 센터이다.
- Centerfor Food Safety and Applied Nutrition CFSAN(식품안전 및 응용영양센터)는 식품과 화장품 산업에 대한 과학적 분석과 연구; 관련 정책의 수립 및 규제를 담당하는 센터이다. CFSAN의 본부는 메릴랜드 주 칼리지 파크 (College Park)에 위치해 있으며, 메릴랜드의 로렐, 일리노이주 베드포드, 알리바바 주의 더플린 아일랜드 등에 분원이 위치해 있다.
- Centerfor Tobacco Products CTP(담배 제품 센터)는 흡연 방지와 담배 규제 법령의 실행을 감독하는 역할을 한다. 이 법규를 관장하는 기관들의 역할은 실행 기준을 정하고, 신규, 일반 담배 제품에 대한 시판전 위험을 평가하고, 위험을 표기하는 라벨링을 감독 관리, 광고와 홍보에 대한 제한을 제시하고 강화하는 역할을 한다.
- Centerfor Veterinary Medicine CVM(수의약품 센터)은 인간과 동물의 건강을 보호하는 것을 목적으로 하는 센터이다. 동물 의약품의 허가를 위해 제품의 안전성과 효과성을 확인하고 관리하는 것을 주목적으로 한다. 본 센터에서는 개, 고양이, 말 등의 애완동물은 물론 돼지, 닭, 꿀벌과 같은 가축들의 의약품 개발 및 승인도 관장한다. 본 센터에서는 고기나 우유, 달걀, 꿀 등 인간에게 먹거리를 제공하는 동물들이 섭취할 수 있는 약물에 대한 안전성도 관리한다. 관리의 대상이 되는 영역은 다음과 같다. - 시판되고 있는 동물 의약품이 안전하고 효과적인지 감시; - 동물, 애완동물 사료, 혹은 간식을 포함한 동물에게 주는 음식이 안전하고, 위생상태가 양호한지, 혹은 적절히 표기가 되었는지 확인; - 동물 사료에 들어간 식품 첨가물이 안전하고 효과적인지 승인 전에 확인; - 동물 의약품, 사료, 및 동물을 사용하여 만든 식품이 안전한지 확인하기 위한 연구 시행; - 물고기, 햄스터, 앵무새 같은 소수 개체의 동물은 물론 개와 가축 등 다수 개체의 동물들을 위해서도 관련 약물의 최소 사용을 위한 의약품 개발 지원
- National Centerfor Toxicological Research NCTR(독성연구센터)은 공공의 건강을 지킨다는 식품의약국의 목적을 달성하기 위해 필수적인 혁신적 정책을 개발하고, 이의 근간이 되는 필수 데이터들을 생산, 제공하기 위한 연구를 수행하는 센터이다. 독성연구 센터 (NCTR) 는 식품의약국 산하 기관 중 유일하에 워싱턴 DC 지역 외부에 소재하고 있으며, 알칸사 주의 제퍼슨에 위치하고 있다.
- Office of Regulatory Affairs 식품의약국의 규제업무총괄실(ORA)은 모든 관련 조직들의 현장 규제업무를 총괄하는 기구이다. ORA 는 규제 대상 제품과 제조 기업을 관리감독하고, 규제 대상 품목의 사례조사를 실시하며 미국에 수입되는 제품들에 대한 심사를 하는 역할을 담당한다. 이를 위해 ORA 는 주정부, 지방 정부기관 및 외국의 기관들과 협업한다.
- Office of Operations 식품의약국의 운영총괄실(OO)은 기관이 비용 및 시간 효과적으로 양질의 서비스를 제공하는 것을 가능하게 만들기 위해 식품의약국과 관련 당국자들에게 운영과 관련된 종합적인 지원을 하는 실조직이다. 업무기획, 분석, 정보보급, 결정 지원 및 기술적 전문성을 통해 식품의약국의 행정 기획과 운영을 체계적으로 지원한다. 임직원 교육 및 관리를 통해 규제 기관의 전반적인 안전 정책과 관련 프로그램의 개발 및 수행을 담당한다.
- 13 개의 실 조직
- Office of the Chief Council
- Office of the Counselor to the Commissioner
- Office of the Executive Secretariat
- Office of Digital Transformation
- Office of Regulatory Affairs
- Office of Clinical Policy & Programs
- Office of External Affairs
- Office of Food Policy & response
- Office of Minority & Health Equity
- Office of Operations
- Office of Policy, Legislation, International Affairs
- Office of the Chief Scientist - Office of the Women’s Health
3. 연방 식품, 의약품 및, 화장품 법령 201(h) 항 소개
- 1938 년 ‘연방 식품, 의약품 및 화장품 법령 (Federal Food, Drug, and Cosmetic Act, 1938)’ 의 제정은 정부의 법제 강화의 일환으로 시행되었으며, 불법 화장품과 의료기기를 포함한 보건의료 품목의 규제 강화를 목적으로 입안되었다.
- 제품이 기기(device)로 분류되기 위해서는 식품, 의약품, 및 화장품 법 201(h) 항의 규정에 따른 의료기기의 개념을 충족해야 한다. 이 법령의 201(h)에 따른 기기(device)란: “도구, 기구, 수단, 기계, 연구, 시약, 혹은 다른 유사한 품목이거나 관련 부품 및 부속물”을 포함한 아래에 해당되는 품목이다.
- 미국 등재 의약품목 (National Formulary)이나 미국 약전(United States Pharmacopoeia), 혹은 그에 준하는 기관이 공식적으로 정한 기준에 포함되거나,
- 사람이나 동물의 질환 진단이나 이와 유사한 상태의 치료 및 증상완화, 처방이나 질환의 방지를 목적으로 사용되거나,
- 사람이나 동물의 신체의 기능을 포함해 어떤 형태로든 영향을 주도록 사용되는 품목 중, 주된 목적이 신체의 화학적 반응을 야기하거나 신진대사에 작용하는 품목이 아닌 것을 의미한다. “기기” 라는 용어에 소프트웨어 기능은 520 (o) 항에 의거하여 제외된다.
4. 2021 주요 FDA 의료기기 가이드라인 변경 사항
- COVID-19 및 보건의료 비상사태 기간에 따른 의료 임상 가이드라인
- 식품의약국( FDA)은 2021년 1월 COVID-19 및 그에 따른 전국적 의료보건 비상사태로 인한 약품 및 의료기기 임상의 개발자와 임상 참가자 그리고 임상 병원 모두의 안전과 원활한 COVID-19 대응 약품 및 의료기기 개발을 위해 신규 가이드라인을 발표하였고 각층의 의견을 수렴하여 2021년 8월 최종 가이드라인이 업데이트 되었다.
- 본 가이드라인을 통해서 FDA는 임상 개발회사들이 임상에 참여하는 모든 인원들에 대한 충분한 COVID19안전대책 마련을 요구하고 있으며 COVID-19 이전 FDA 허가를 받은 임상 IND들도 임상 프로토콜 변경 등을 통해 실제 임상 진행이 COVID-19로 지연되거나 임상참가자들 사이에서 COVID-19가 전파되는 것을 방지하도록 명시하고 있다.
- 또 한 실제 COVID-19의 영향으로 임상진행에 문제가 발생한 다양한 실제 사례들에 대한 답변을 가이드라인에 포함시켜 산업 전반에 명확한 메세지 전달과 함께 FDA가 COVID-19 방지를 위한 신규 가이드라인 아래 임상시험 허가 및 모니터링을 하고 있음을 알 수 있다.
- FDA 의료기기 실행 계획으로서 인공지능/머신러닝 기반 소프트웨어 가이드라인 (2021 년 3월 12 일)
- 2021 1월 12일, 미국 FDA 는 의료기기 실행 계획으로서 인공지능 및 머신러닝 기반 소프트웨어 가이드라인 (Artificial Intelligence/Machine Learning (AI/ML)
- Based Software as a Medical Device (SaMD) Action Plan, 2021.1) 을 발표했다. 실행 계획(Action Plan) 은 2019 년 4월 토론 문서인 “의료기기로서 인공지능/머신러닝 기반 소프트웨어 수정을 위해 제안된 규제 프레임워크 (Proposed Regulatory framework for Modification to Artificial Intelligence/Machine Learning-Based Software as a Medical Device)” 문서의 이해관계자 피드백에 대한 FDA 의 직접적인 응답이고, 주요 내용은 아래와 같다:
- 인공지능/머신러닝 기반 SaMD에 맞춤화된 규제 프레임 워크’로 업데이트 실시
- Good Machine Learning Practice (GMLP) 개발 촉진
- 사용자에게 투명성을 제공하는 환자 중심 접근방식
- 알고리즘 편향 식별, 제거 및 견고성과 관련된 규제과학 방법론 개발
- 인공지능/머신러닝 기반 SaMD를 위한 Real-World Performance (RWP) 프로세스를 시험하는 관계자와의 협력
LIST
'바이오메디컬' 카테고리의 다른 글
| FDA - OTC 기기 (0) | 2022.10.28 |
|---|---|
| 식약처 ‘생산·수입 중단 보고대상 의료기기’ 1,516개 제품 지정 공고 (0) | 2022.10.26 |
| 규제 장벽 ‘의료기기’…빠른 상용화 위한 제도 개편 시급 (0) | 2022.10.26 |
